Malaysia's medical device market continues to grow rapidly, attracting both local manufacturers and international brands. However, before any device can be legally marketed, it must comply with Malaysia's Medical Device Authority (MDA) requirements.
A crucial first step in the registration journey is understanding medical device classification in Malaysia. Classification determines the regulatory pathway, documentation requirements, and approval timeline.
This guide explains everything you need to know about medical device regulation and classification in Malaysia. For a full walkthrough of the registration process, see our guide on how to register a medical device in Malaysia.
Key Takeaways
- Malaysia uses a risk-based classification system (Class A to D).
- Correct classification is critical for registration success.
- Higher risk devices require more documentation and scrutiny.
- CAB assessment is required for Class B, C, and D devices.
- Early regulatory planning reduces approval delays.
Why Medical Device Classification Matters
Medical device classification determines the entire regulatory pathway for your product. Getting it right from the start is the single most important step before applying for registration.
Incorrect classification is one of the most common reasons for delays or rejection during product registration.
Regulatory FrameworkOverview of Medical Device Regulation in Malaysia
Malaysia regulates medical devices under the Medical Device Act 2012 (Act 737) and the Medical Device Regulations 2012. The Medical Device Authority (MDA) oversees device registration, establishment licensing, post-market surveillance, and import and distribution compliance.
All medical devices must be registered with the MDA before they can be imported, manufactured, distributed, or sold in Malaysia.
Classification SystemMalaysia's Risk-Based Classification System
Malaysia follows a risk-based classification system aligned with the Global Harmonisation Task Force (GHTF) model. Medical devices are divided into four classes — the higher the risk, the stricter the regulatory requirements.
| Class | Risk Level | Regulatory Control |
|---|---|---|
| Class A | Low risk | Minimal regulatory control |
| Class B | Low to moderate risk | Moderate control |
| Class C | Moderate to high risk | High control |
| Class D | High risk | Very high control |
Medical Device Classification: Class A to D Explained
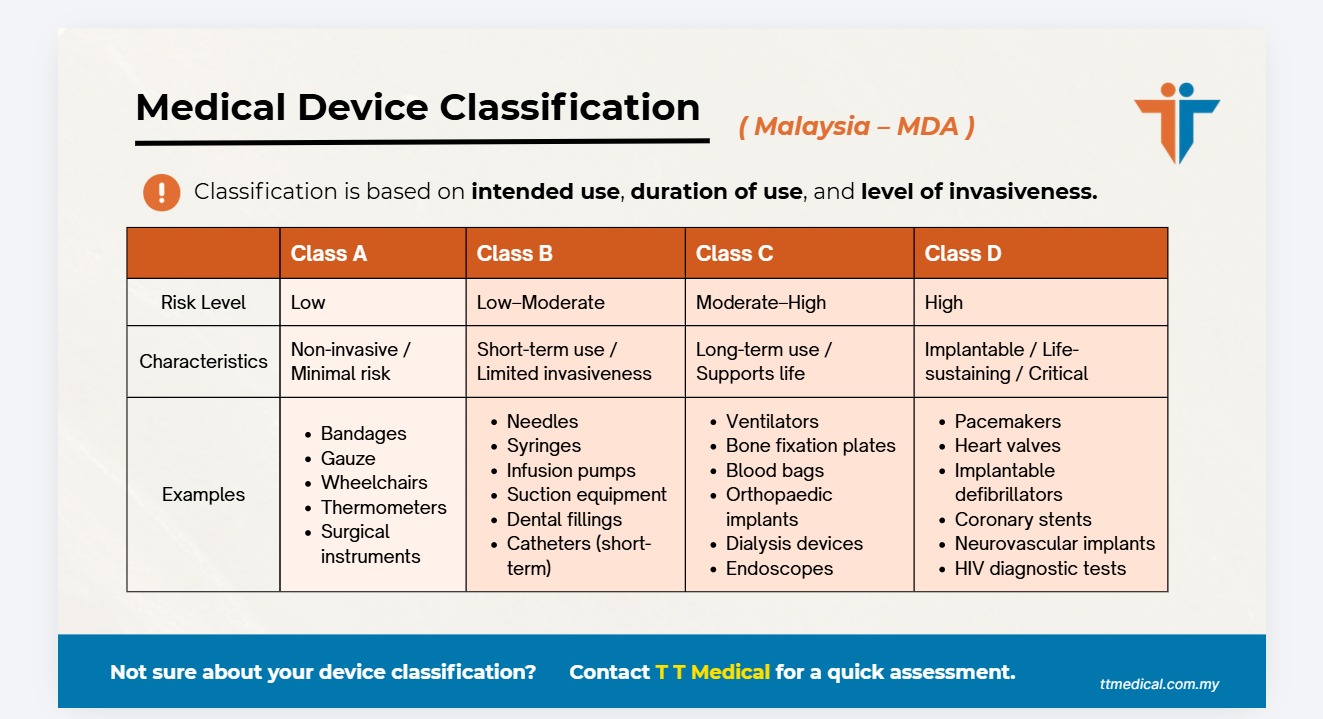
Wheelchairs, surgical retractors, non-sterile bandages, hospital beds.
- Simplified registration pathway
- Lighter conformity assessment
- Shorter approval timeline
Hypodermic needles, suction equipment, infusion pumps, dental fillings.
- CAB review required
- Technical documentation needed
- Moderate registration timeline
Ventilators, bone fixation plates, blood bags, orthopaedic implants.
- Comprehensive technical documentation required
- Clinical evidence often necessary
- Longer review and approval process
Heart valves, implantable defibrillators, HIV diagnostic tests, implantable pacemakers.
- Full conformity assessment required
- Extensive clinical data needed
- Strict post-market surveillance obligations
How Devices Are Classified: The Key Rules
The MDA uses classification rules based on four main factors. Each one directly influences which class your device falls into.
What the manufacturer claims the device is designed to do — whether for diagnosis, monitoring, treatment, or life support. This is the most important factor in classification. Two similar-looking products can fall into different classes based solely on how their intended use is described.
Devices are categorised by how long they interact with the body: transient (less than 60 minutes), short-term (up to 30 days), or long-term (more than 30 days). Longer contact generally means a higher risk classification.
Risk increases based on how the device interacts with the body — from non-invasive (bandages, stethoscopes) to invasive via body orifice (catheters, endoscopes), surgically invasive (bone plates, sutures), and implantable (pacemakers, joint replacements). Implantable devices typically fall under Class C or D.
Active devices rely on electricity or an external power source and are frequently placed in higher risk classes due to the greater potential for harm from malfunction. Non-active devices operate mechanically and often fall in lower classes — though implantable non-active devices can still be Class C or D. Learn more in our guide on active vs non-active medical devices.
In Vitro Diagnostic (IVD) Devices
Malaysia's medical device regulation also includes a dedicated classification system for In Vitro Diagnostic (IVD) devices, which are used to test human samples such as blood or urine.
These devices support disease detection, screening, monitoring, and treatment decisions. Common examples include pregnancy tests, glucose meters, COVID-19 test kits, and laboratory reagents.
IVD devices are classified based on the potential public health impact of incorrect results. Tests used to screen blood donations or detect infectious diseases are considered higher risk compared to routine or self-testing kits.
Higher-risk IVDs must provide strong performance data, validation studies, and quality system certification before registration.
Role of Conformity Assessment Bodies (CAB)
Conformity Assessment Bodies (CABs) play a critical role in Malaysia's regulatory approval process for Class B, C, and D devices.
CABs are authorised by the MDA to evaluate whether a device meets safety, quality, and performance requirements before submission.
Quality management systems (ISO 13485), technical documentation, risk management, product testing, and clinical evidence are all assessed during the conformity review process.
Steps After Classification: The Registration Pathway
Once classification is confirmed, the full registration process begins. Proper classification ensures a smoother and faster regulatory journey.
Foreign manufacturers must appoint a LAR in Malaysia to handle all regulatory submissions and MDA communications.
Class B, C, and D devices must undergo CAB review before submission to the MDA.
Technical documentation must be compiled in the CSDT format as required by the MDA.
The LAR submits the completed application through the MDA's official online system.
Once approved, the device is listed in the MDA system and can legally be imported and distributed in Malaysia.
Common Mistakes in Medical Device Classification
Many companies experience delays due to avoidable classification errors. These are the most frequent mistakes we see during the registration process.
Final Thoughts
Successfully navigating medical device classification in Malaysia is the foundation of a smooth and timely registration process. Getting the classification right from the start helps avoid costly delays, reduces regulatory risks, and ensures your device aligns with Malaysia's requirements.
If you're planning to register a medical device in Malaysia, early classification is the first and most important step. TT Medical Management supports companies with device classification assessment, regulatory strategy planning, CSDT dossier preparation, MDA submission, and Local Authorised Representative services.
FAQ