
A Declaration of Conformity (DoC) is a mandatory document in which the manufacturer formally declares that their medical device complies with all applicable Essential Principles of Safety and Performance (EPSP) under Malaysia's Medical Device Act 2012 (Act 737) and the Medical Device Regulations 2012. It must be prepared on the manufacturer's letterhead, signed by company top management, and submitted as part of every MDA registration application — for all device classes from A to D.
Every medical device registration application submitted to Malaysia's Medical Device Authority (MDA) requires a Declaration of Conformity. Yet it's one of the most frequently misunderstood documents in the submission package — either drafted incorrectly, signed by the wrong person, or confused with the EU Declaration of Conformity used in international submissions.
The Malaysian DoC is a specific, legally required document governed by its own MDA guidance. Getting it wrong — even with an otherwise complete technical file — can delay your submission. This guide explains exactly what a DoC is, what it must contain, who must sign it, and the common mistakes that cause rejections.
The DoC sits within the broader documentation framework for medical device registration in Malaysia. For a full picture of all required documents, see our complete MDA document checklist.
Key Takeaways
- The DoC is required for all device classes — A, B, C, and D — without exception.
- It must be prepared on the manufacturer's official letterhead and signed by top management.
- The MDA has its own DoC format — it is not the same as an EU Declaration of Conformity.
- The DoC must reference the specific device, its classification, the applicable EPSPs, and the standards used to demonstrate conformity.
- For re-registration, an updated DoC must be submitted — a DoC from the original application is not sufficient.
- A DoC that is unsigned, undated, incorrectly formatted, or signed by someone other than top management will be rejected.
What Is a Declaration of Conformity (DoC)?
A Declaration of Conformity is a formal written attestation drawn up by the manufacturer stating that their medical device fully complies with all applicable Essential Principles of Safety and Performance (EPSP) and all other requirements of the Medical Device Act 2012 (Act 737) and the Medical Device Regulations 2012.
In simple terms: the manufacturer is declaring, in a legally binding document, that they have assessed their device against every applicable requirement and that the device meets all of them. It is not a third-party certificate — it is the manufacturer's own attestation. The MDA accepts it as such, but requires it to be supported by sufficient evidence across the rest of the technical documentation.
The legal basis for the DoC is Item 9 of the Third Schedule of the Medical Device Regulations 2012, which requires every manufacturer to draw up a written Declaration of Conformity and provide sufficient documents or evidence to support it.
Who Needs a DoC and When
Unlike some other documentation requirements that scale with device classification, the DoC is required for every device regardless of risk class. Class A, B, C, and D devices all need a DoC as part of the registration submission. There is no pathway or exemption that removes this requirement.
The DoC must be prepared and submitted as part of the initial MDA registration application via MeDC@St. It must be current — dated at or close to the time of submission — and must accurately reflect the device as described in the rest of the technical file.
For re-registration, an updated DoC must be provided. The original DoC from a previous registration cycle is not sufficient. Per MDA/GD/0070, the DoC must be reviewed and updated to reflect the current state of the device and its compliance with all applicable requirements at the time of re-submission.
If a significant change is made to the device after registration — such as a design modification, change of manufacturing site, or update to the intended use — a revised DoC may need to be submitted as part of the change notification process to the MDA.
What Must the DoC Contain?
The MDA prescribes a specific format for the DoC in Appendix 1A of the Medical Device Regulations 2012. Every DoC must include all of the following elements. Missing any of them is a common reason for rejection.
| Manufacturer details | Full legal name, registered address, and contact details of the device manufacturer |
| Device identification | Device name, model number(s), catalogue number(s), and all configurations covered by the declaration |
| Device classification | The risk class of the device (Class A, B, C, or D) under Malaysian classification rules |
| Intended use statement | The intended use of the device as stated in the CSDT and labelling — must be consistent throughout the entire submission |
| EPSP conformity statement | A declaration that the device conforms to all applicable Essential Principles of Safety and Performance under Act 737 |
| Standards applied | Reference to the specific standards used to demonstrate conformity with each applicable EPSP (e.g. ISO 14971, IEC 60601) |
| Supporting documentation | Reference to the technical documentation that supports the declaration — typically the CSDT and associated test reports |
| Date and place | The date the DoC was drawn up and the place of issue |
| Authorised signatory | Name, title, and signature of the company's top management authorised to sign on behalf of the manufacturer |
| Manufacturer letterhead | The DoC must be prepared on the manufacturer's official letterhead — not a blank template or third-party document |
Who Must Sign the Declaration of Conformity?
This is one of the most common points of confusion. The MDA is specific: the DoC must be signed by the manufacturer's top management — not a regulatory affairs manager, not a quality engineer, and not a member of staff acting without proper authorisation.
Typically the CEO, Managing Director, President, or a person in an equivalent position who has the legal authority to bind the company. Their title and full name must be stated on the DoC alongside their signature.
A Regulatory Affairs Manager, QA Manager, or any other staff member below top management level cannot sign the DoC unless they hold a formal written delegation of authority from top management. Even then, the delegation document may be scrutinised.
The Local Authorised Representative is responsible for submitting the DoC but cannot draw it up or sign it. The DoC is the manufacturer's own attestation — only the manufacturer can make it. The LAR submits it as part of the MeDC@St application package.
The signatory's name and title on the DoC must be consistent with the manufacturer information provided elsewhere in the CSDT and MeDC@St application. Discrepancies in name, title, or company details will be queried by the MDA.
Malaysian DoC vs EU Declaration of Conformity
Manufacturers with CE marking often ask whether their existing EU Declaration of Conformity can be used for the Malaysian MDA submission. The answer is: not as a substitute — but it can be submitted alongside the MDA-format DoC as supporting evidence.
| Legal basis | MDA: Act 737 and Medical Device Regulations 2012 | EU: EU MDR 2017/745 or IVDR 2017/746 |
| EPSP references | MDA: Malaysian EPSP framework | EU: General Safety and Performance Requirements (GSPR) under MDR |
| Format required | MDA: Appendix 1A format (MDR 2012) — mandatory | EU: EU MDR Annex IV format |
| Accepted as substitute | No — the MDA requires its own format. An EU DoC cannot replace it |
| Can be submitted together | Yes — submitting both is acceptable and may support the submission as additional evidence |
| Regulatory authority | MDA DoC is for MDA submission only. EU DoC is for European regulators only |
Common DoC Mistakes That Delay Submissions
The most frequent mistake. Manufacturers with CE marking assume their EU DoC satisfies the Malaysian requirement. The MDA will reject any DoC that does not follow the Appendix 1A format prescribed in the Medical Device Regulations 2012.
DoCs signed by a Regulatory Affairs Manager, QA Director, or any person without clear top management authority will be queried. The signatory must be a person who can legally bind the manufacturer organisation.
The intended use stated in the DoC must be word-for-word consistent with the intended use in every other part of the submission — CSDT, IFU, labelling, and MeDC@St form. Any inconsistency triggers an MDA query.
The DoC must reference the specific standards applied to demonstrate conformity with each applicable EPSP. Vague statements like "complies with applicable standards" without naming the standards are not acceptable.
Submitting the original DoC from a previous registration cycle without updating it is a common error at renewal. An updated DoC is required for every re-registration submission, reflecting the current device and its compliance status.
The DoC must be prepared on the official company letterhead of the manufacturer — not a generic template, not the LAR's letterhead, and not a blank document. The letterhead establishes that this is an official company declaration.
Where the DoC Sits in Your Full Submission
The DoC is one part of a complete submission package. Understanding how it relates to the other documents helps ensure everything is consistent and the MDA review proceeds without queries.
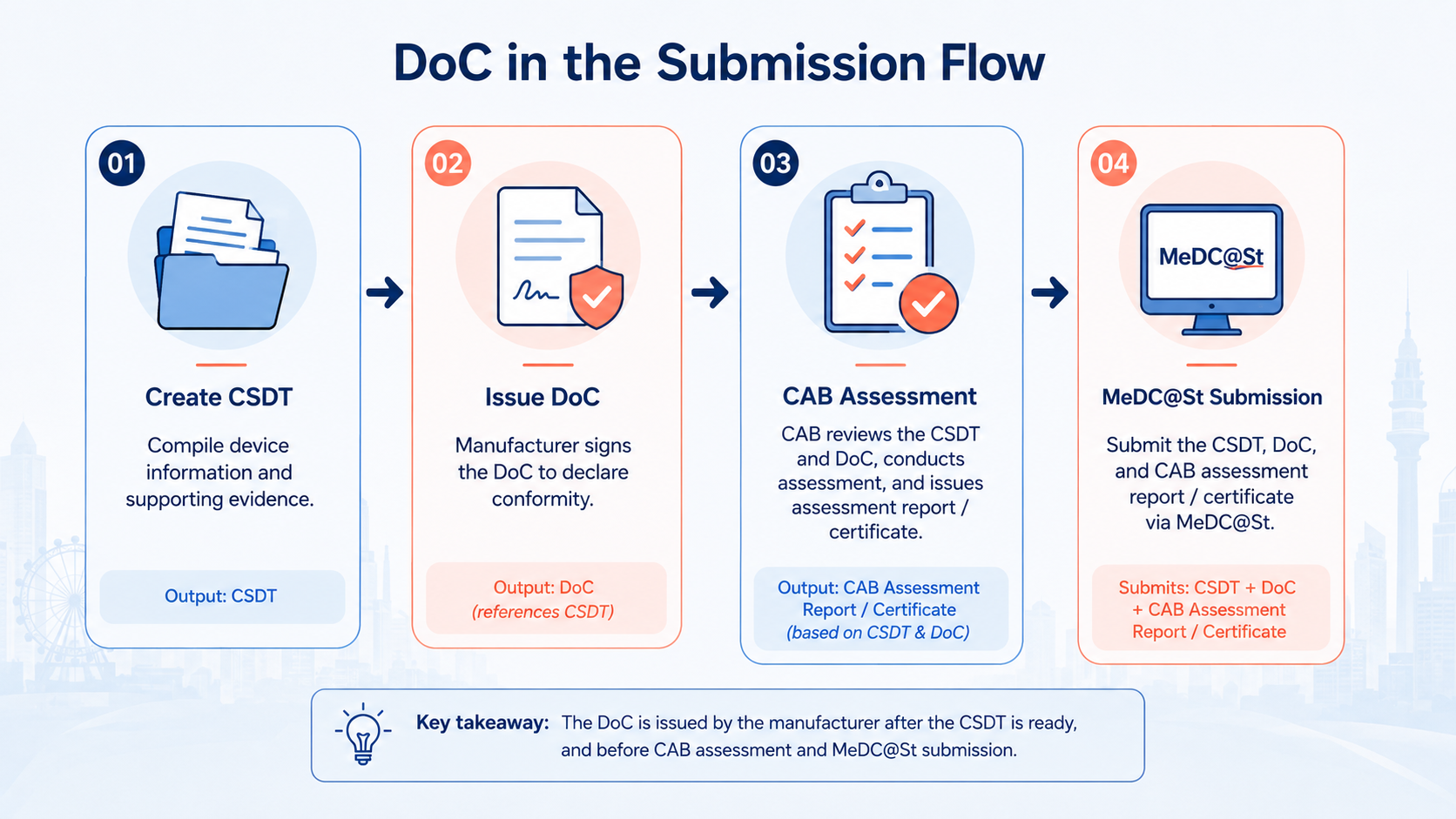
The DoC declares that the device meets all applicable EPSPs. The ERSP checklist in the CSDT demonstrates how each EPSP is met, with cross-references to supporting documents. The two must be consistent — EPSPs referenced in the DoC must appear in the checklist, and vice versa.
The DoC is separate from the CSDT but closely linked to it. The CSDT provides the evidence that supports the declaration made in the DoC. Statements in the DoC — particularly the intended use and standards applied — must match what is stated throughout the CSDT. See our full MDA document checklist for how the DoC fits into the complete submission package.
The LAR submits the DoC as part of the MeDC@St application but does not draw it up or sign it. The DoC must come directly from the manufacturer — it is the manufacturer's legal attestation, not the LAR's. The LAR's role is to review it for completeness and consistency before submission.
For Class B, C, and D devices, the DoC is reviewed as part of the conformity assessment conducted by the registered CAB. The CAB checks that the DoC is correctly formatted, properly signed, and consistent with the rest of the technical documentation before issuing its certificate.
DoC Review and Submission Support from TT Medical
Getting the DoC right is straightforward once you know exactly what the MDA requires — but the details matter. TT Medical reviews every DoC before it goes to the CAB or MDA, checking format, signatory authority, consistency with the CSDT, and standards references.
Final Thoughts
The Declaration of Conformity is not a formality — it is a legally binding attestation that sits at the heart of your MDA registration. Getting it right means using the correct MDA format, ensuring it is signed by top management, keeping it consistent with every other document in your submission, and updating it for every re-registration cycle.
If your DoC is rejected or queried, the rest of your submission stalls while corrections are made. The good news is that DoC errors are entirely avoidable with the right preparation. If you are unsure whether your DoC meets MDA requirements, speak to our consultancy team before submitting.
FAQ