
A Conformity Assessment Body (CAB) is an independent organisation registered by Malaysia's Medical Device Authority (MDA) under the Medical Device Act 2012 (Act 737) to evaluate medical devices and quality management systems for regulatory compliance. For Class B, C, and D devices, a CAB must assess your technical documentation and issue a certificate before you can submit your registration application to the MDA. Class A devices are exempt from CAB assessment.
If you're preparing to register a medical device in Malaysia, the CAB is the organisation that stands between your technical file and your MDA submission. For most manufacturers, it's the most intensive step in the registration process — and the one where delays most commonly occur.
Understanding what a CAB actually does, how it assesses your submission, and what it's looking for is the most effective way to prepare a dossier that gets through cleanly. This guide covers the CAB's role in full — what it is, what it reviews, how the two assessment routes work, and how to choose the right CAB for your device.
Key Takeaways
- A CAB is an independent body registered by the MDA to assess medical devices and quality management systems for compliance with Act 737.
- Class B, C, and D devices must be assessed by a registered CAB before the manufacturer can submit to the MDA — this is not optional.
- Class A devices are exempt from CAB assessment under the Medical Device (Exemption) Order 2024.
- There are two CAB assessment routes: full assessment (for devices without prior international approval) and verification (expedited, for devices approved by a recognised authority).
- The CAB reviews technical documentation, quality management systems, labelling, clinical evidence, and the Declaration of Conformity.
- The CAB must be registered with the MDA and have expertise in the relevant Medical Device Technical Area (MDTA) for your device.
- If the CAB finds non-conformities, the application can be rejected before it ever reaches the MDA.
What Is a Conformity Assessment Body (CAB)?
A Conformity Assessment Body is an independent organisation that has been evaluated, approved, and registered by the Medical Device Authority (MDA) to conduct conformity assessments of medical devices and establishments in Malaysia. The legal basis for CABs is Section 7 of the Medical Device Act 2012 (Act 737), which mandates that medical devices undergo a conformity assessment process before registration.
The CAB's function is to act as an independent technical reviewer — verifying that a device meets the Essential Principles of Safety and Performance (EPSP), that the manufacturer's quality management system is adequate, and that the technical documentation submitted is complete and consistent. Only after the CAB issues a favourable assessment report and certificate can the registration application proceed to the MDA.
What Does a CAB Assess?
The CAB's assessment is systematic and covers multiple areas of the registration dossier. For device registration, the CAB evaluates the following as a minimum.
The CAB reviews the full Common Submission Dossier Template (CSDT) including device description, design verification and validation, clinical evidence summary, labelling, and manufacturer information. Every referenced supporting document must be submitted in full.
The CAB verifies that each applicable EPSP has been addressed in the checklist, with the correct standards referenced (full title, number, date, and issuing body) and corresponding controlled documents cross-referenced throughout the CSDT.
The CAB verifies the manufacturer's QMS certification — typically ISO 13485 — for authenticity and validity. Both vertical standards (ISO 13485) and horizontal standards relevant to the device type must be stated in the Declaration of Conformity.
The CAB reviews the DoC for correct format (Appendix 1A), proper signatory authority (top management), consistency with the CSDT intended use, and confirmation that the device conforms to all applicable EPSPs. See our DoC guide for full requirements.
The CAB verifies that the risk classification and classification rule applied are correct under the First Schedule of the Medical Device Regulations 2012 and the relevant MDA guidance documents. If the CAB determines the classification is incorrect, it will flag this before proceeding.
All labels, packaging, and Instructions for Use are reviewed for consistency with the intended use, CSDT content, and clinical evidence. Labelling must meet MDA requirements including brand name, GMDN code, and for home-use devices, Bahasa Malaysia translation.
Full Assessment vs Verification: The Two CAB Routes
How a CAB assesses your device depends on whether it has prior approval from a recognised international regulatory authority. There are two distinct routes.
| Factor | Full Assessment | Verification Route |
|---|---|---|
| Eligibility | Devices without prior approval from a recognised authority | Devices approved by FDA, EU CE, HSA, TGA, Health Canada, or Japan MHLW (per MDA Circular Letter No. 1 Year 2025) |
| CAB approach | Full independent technical review of all documentation | CAB relies on prior international assessment — focuses on verification of that prior approval |
| CAB review time | Longer — full technical assessment of every element | 30 working days (target for verification route) |
| Documents required | Full CSDT plus all supporting documents | Same core documents, plus copies of reference country approval certificates |
| Outcome | CAB certificate and conformity assessment report | CAB verification certificate and assessment report |
| What goes to MDA | Full CSDT + CAB certificate and report | Full CSDT + CAB verification certificate and report |

How the CAB Assessment Process Works
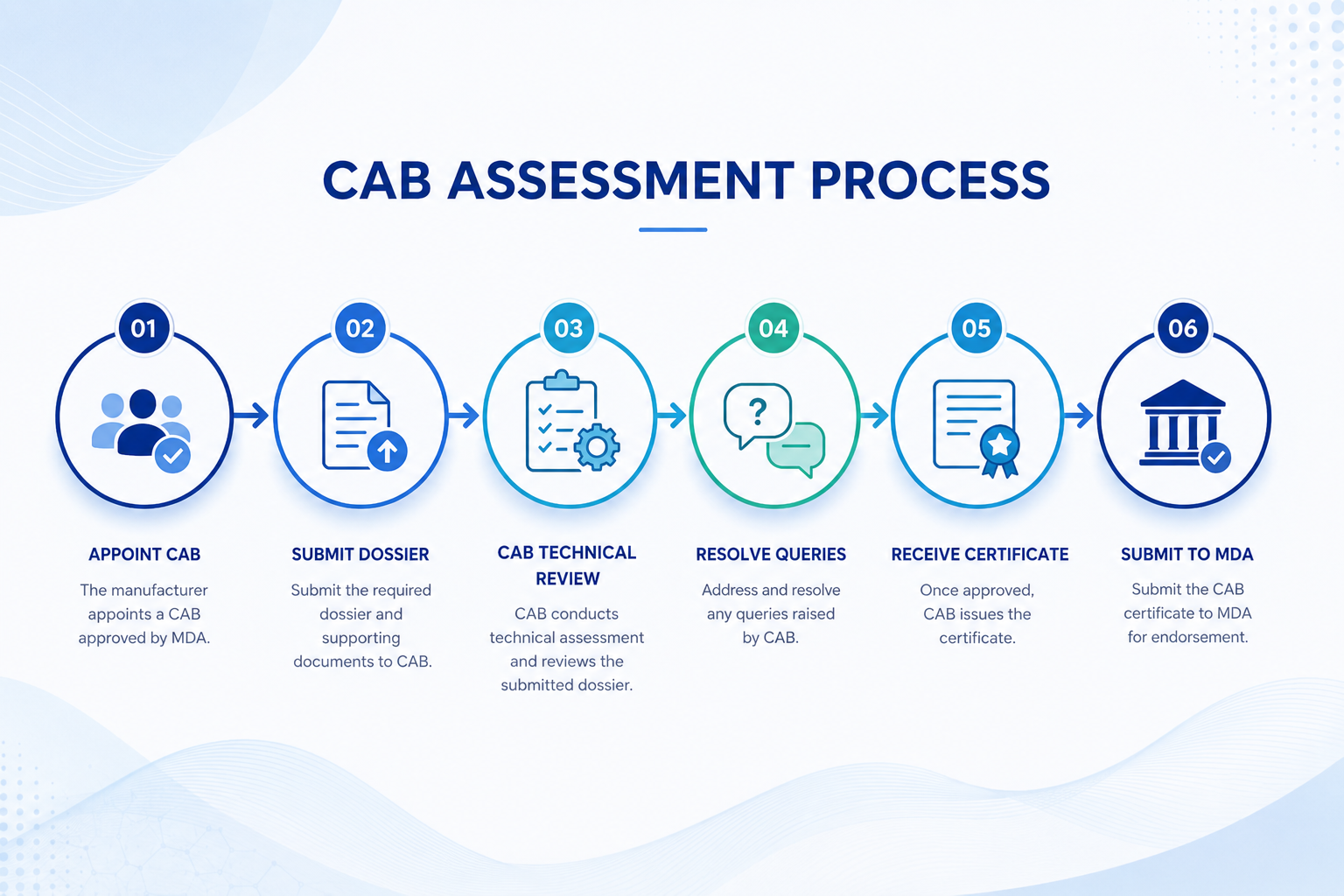
The manufacturer (or their LAR) appoints a CAB that is registered with the MDA and holds expertise in the relevant Medical Device Technical Area (MDTA) for your device. The CAB must have the right technical scope — not all CABs assess all device types.
The complete CSDT, Declaration of Conformity, ISO 13485 certificate, labelling, and all supporting documents are submitted to the CAB. Every referenced document must be included in full. Partial submissions will result in queries and delays.
The CAB's technical personnel review the dossier systematically — verifying classification, EPSP conformity, QMS certification, clinical evidence, labelling, and DoC. The target review time set by MDA guidance is 4 hours per device application for report writing and certificate issuance.
If the CAB identifies non-conformities or missing information, it raises queries with the manufacturer or LAR. These must be addressed before the assessment can proceed. Non-conformities found by the CAB can result in rejection of the application before it reaches the MDA.
A successful assessment results in a signed CAB certificate of conformity and a full assessment report. Both documents are mandatory inclusions in the MDA submission package — the certificate must state the CAB name, expiry date, and assessment route (full or verification).
The same technical dossier reviewed by the CAB, together with the CAB certificate and assessment report, is submitted to the MDA via MeDC@St. The MDA targets 60 working days for Class B, C, and D devices. Because the CAB has comprehensively pre-reviewed the file, MDA review is typically faster than the full timeline suggests.
How to Choose the Right CAB for Your Device
Not every registered CAB is authorised to assess every type of device. Choosing the wrong CAB — one without the right MDTA scope — will result in the assessment being invalid for MDA submission. Here is what to check before appointing a CAB.
The CAB must be currently registered with the MDA. Verify their registration on the MDA's official list of registered CABs. An assessment from an unregistered body is invalid.
Each CAB is registered with a specific scope covering defined Medical Device Technical Areas. Confirm that the CAB's registered MDTA scope covers your device category before submitting — not all CABs can assess all device types.
A CAB experienced in Class C or D device assessment, or in the verification route for your specific reference country, will be more efficient and less likely to raise unnecessary queries. Ask about their track record with devices similar to yours.
The MDA requires CABs to maintain documented procedures for independence and impartiality, including conflict-of-interest management. This is particularly relevant if the CAB is a subsidiary of a foreign notified body — procedures must be in place to separate the verification personnel from the original assessment personnel.
The MDA targets 4 hours per device application for CAB assessment, but the practical timeline from submission to certificate varies depending on CAB workload and query volume. Clarify expected turnaround and communicate timelines with your LAR before appointment.
CAB vs MDA: Understanding the Difference
An independent, MDA-registered organisation that conducts the technical review of your device dossier, verifies QMS certification, and issues a certificate and assessment report. It is a private entity — not a government body — but it operates under MDA authorisation and oversight. Its assessment is the essential prerequisite for MDA submission.
The government regulatory authority under Malaysia's Ministry of Health, responsible for implementing Act 737, maintaining the medical device register, and issuing registration certificates. The MDA receives your application after the CAB has completed its assessment — it does not conduct its own full technical review from scratch, as the CAB has already done this.
What Happens If the CAB Finds Non-Conformities?
If the CAB identifies issues in the dossier — whether in the technical documentation, QMS certification, labelling consistency, or DoC — it will raise findings that must be resolved before the assessment can be completed. In serious cases, the application can be rejected entirely before it reaches the MDA.
Requests for clarification, additional documentation, or corrections to specific inconsistencies. These extend the timeline but can typically be resolved without restarting the assessment.
Substantive issues with the technical documentation, QMS evidence, or classification — such as an incorrect EPSP checklist, missing test reports, or inadequate clinical evidence for the device class. These require more significant rework.
If non-conformities in the QMS, post-market surveillance system, or complete device dossier are significant enough, the CAB may reject the application. A rejected application must be corrected and resubmitted — resetting the assessment timeline entirely.
The best way to avoid CAB rejection is to prepare documentation to CAB-ready standard before submission — not MDA-ready standard. A clean first submission to the CAB is what protects your overall registration timeline. If you're unsure whether your dossier is ready, our team can review it before it goes to the CAB.
How We Can HelpCAB Coordination and Submission Support from TT Medical
TT Medical manages the CAB coordination process on behalf of manufacturers as part of our full registration service. This means selecting the right registered CAB for your device type, preparing the dossier to the standard required for a clean first submission, managing CAB queries, and then handling the full MDA submission once the CAB certificate is issued.
Final Thoughts
The CAB is the most intensive quality gate in Malaysia's medical device registration process. Getting through it cleanly — with a complete, consistent, well-structured dossier — is the single most important factor in protecting your registration timeline.
Manufacturers who treat the CAB stage as an internal review round before the "real" submission to the MDA consistently achieve faster approvals than those who submit to the CAB while still finalising their documentation. Prepare to CAB standard first. If you need support with that process — from CSDT preparation through to CAB appointment and query management — speak to our consultancy team.
FAQ