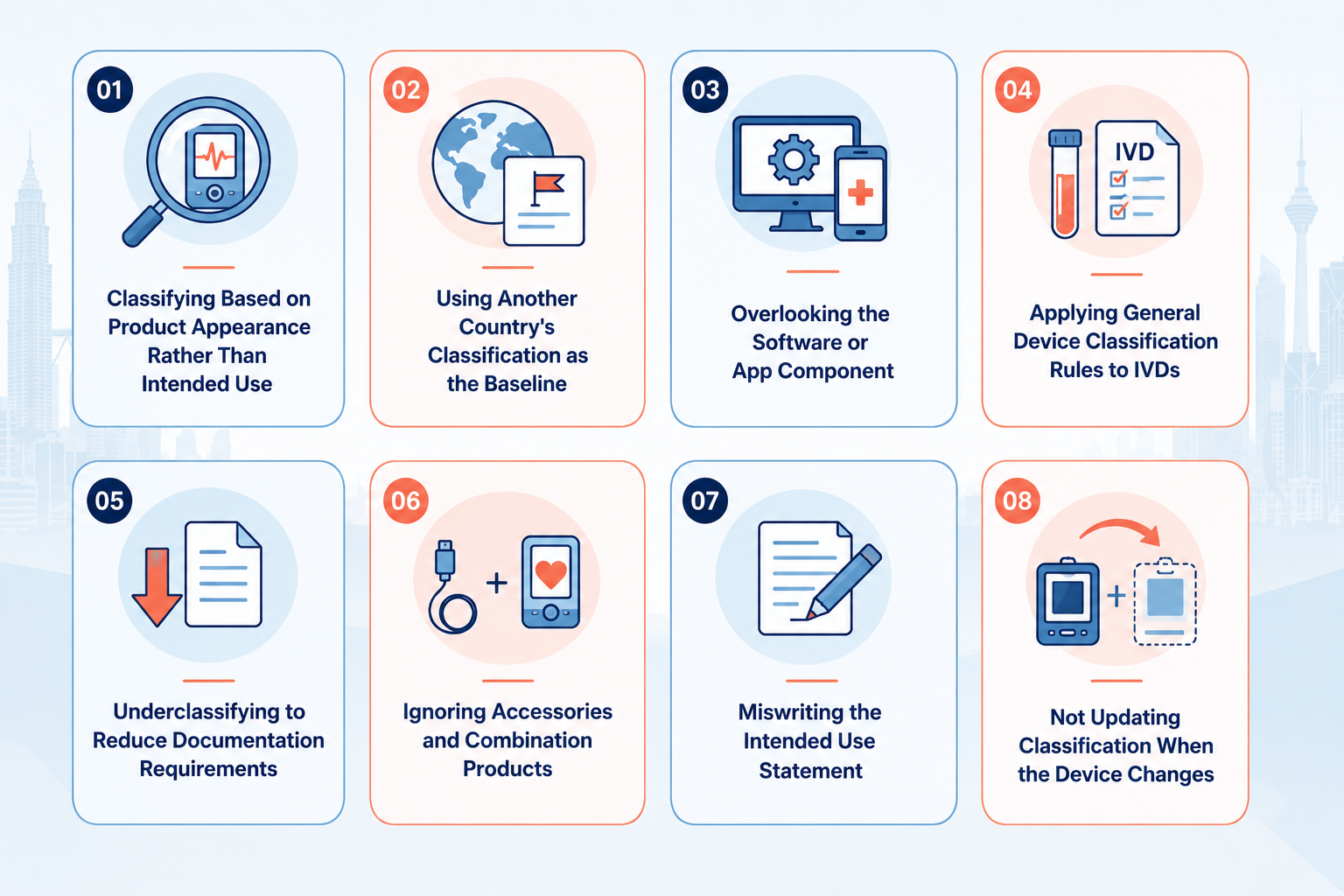
The most common medical device classification mistakes in Malaysia include misclassifying based on product appearance rather than intended use, applying general device rules to IVDs, overlooking software components, and submitting without first validating classification with the MDA. Each error can result in reclassification requests, additional documentation requirements, and delays of several months.
Classification is the starting point for everything in medical device registration in Malaysia. It determines which documents you need, whether a conformity assessment is required, and how long the MDA review process will take. Get it right, and the path ahead is clear. Get it wrong, and you may not find out until the MDA sends your submission back.
The Medical Device Authority (MDA) classifies devices based on risk, not product category or marketing label. This distinction is where most mistakes happen. Manufacturers often approach classification the way they would classify their own products for commercial purposes — which is not the same as how the MDA evaluates them.
This guide covers the most common classification mistakes we see during the registration process, why they happen, and what to do instead. If you're new to the topic, it helps to read our guide on the medical device classification framework in Malaysia first.
Key Takeaways
- Classification must be based on intended use and how the device achieves its medical purpose — not how it looks or how it is marketed.
- Two physically identical products can fall into different classes depending on their stated intended use.
- IVDs have their own classification rules that are separate from general medical device rules.
- Software components, accessories, and companion products each require their own classification assessment.
- Misclassification discovered during MDA review can reset the entire submission timeline.
- Getting classification confirmed before preparing documentation is always faster and less costly than correcting it later.
 Mistake 1
Mistake 1
Classifying Based on Product Appearance Rather Than Intended Use
This is the most common classification mistake. Manufacturers look at their product, compare it to similar-looking devices already on the market, and assume they share the same class. In many cases, they don't.
The MDA classifies devices based on their intended use — specifically, how the device achieves its medical purpose, who it is used on, and what happens if it malfunctions or produces an incorrect result. Two products with identical physical forms can fall into completely different classes based on their intended use statement alone.
Example: A standard bandage used for wound covering is Class A. The same bandage, if it incorporates an antimicrobial agent and claims to prevent infection, may move into Class B. The physical product is nearly identical — the classification is determined entirely by what the manufacturer claims the device does.
Using Another Country's Classification as the Baseline
A device classified as Class II under US FDA rules or Class IIa under the EU MDR is not automatically Class B in Malaysia — even though the risk levels appear comparable. Malaysia uses its own classification rules under the Medical Device Act 2012 (Act 737), aligned with AMDD guidelines but interpreted and applied by the MDA independently.
Submitting with a classification based on your home market approval — without verifying it against Malaysian rules — is one of the most avoidable causes of reclassification requests. The MDA will assess your device against its own criteria, regardless of what class has been assigned elsewhere.
Prior international approvals are useful as supporting evidence in your technical file. They are not a substitute for confirming the correct Malaysian classification before you begin.
Overlooking the Software or App Component
A physical device and the software that drives it are not always the same regulatory entity. If a device includes embedded software — or if there is a companion app that performs a diagnostic, monitoring, or clinical decision support function — that software component may itself qualify as a medical device under Malaysian regulations.
Software as a Medical Device (SaMD) is treated as an active medical device by the MDA, even if it runs on a general-purpose phone or laptop. If your hardware is Class A or B but the companion app makes clinical claims, the software may require its own separate registration — and may fall into a higher class than the hardware.
This is particularly common with connected health devices, wearables, and diagnostic platforms where the clinical function is performed by the software, not the physical sensor alone.
Applying General Device Classification Rules to IVDs
In Vitro Diagnostic devices have their own classification rules that are entirely separate from general medical device classification. General devices are classified based on how they interact with the body — invasiveness, duration of use, active versus non-active. IVDs do not interact directly with the patient's body during the test, so these rules do not apply.
IVDs are classified based on the risk of an incorrect test result — specifically the potential public health impact and clinical consequences of a false positive or false negative. A blood glucose meter looks like a simple device, but it is not classified the same way as a bandage of equivalent physical simplicity. Learn more in our guide to IVD device classification in Malaysia.
Underclassifying to Reduce Documentation Requirements
The appeal of a lower classification is understandable — fewer documents, no conformity assessment, faster approval. But deliberately underclassifying a device — or framing the intended use in a way that obscures the true risk level — is not a viable strategy.
The MDA reviews submissions based on the actual device and its realistic use in the Malaysian healthcare context. If the device does not match the classification stated in the submission, the MDA will issue a reclassification request. This typically requires a fresh submission with revised documentation aligned to the correct class — which costs significantly more time and money than preparing the right documentation from the start.
Ignoring Accessories and Combination Products
Accessories to medical devices — such as replacement sensors, dedicated carrying cases with clinical function, or add-on components — are not automatically covered under the parent device's registration. Depending on their function, accessories may require their own classification and registration with the MDA.
Similarly, combination products — devices that incorporate a drug component, or products where a physical device and a pharmaceutical are sold together — require careful assessment of which function is primary. The primary function determines the applicable classification rules and whether the product falls under MDA, NPRA, or requires review by the Medical Device-Drug-Cosmetic Interphase Committee (MDDCI). Refer to our guide on the difference between MDA and NPRA for more on this.
Miswriting the Intended Use Statement
The intended use statement is the single most important document in your classification decision. It defines what your device is, what it does, and who it is used on. The MDA uses this statement — not your product brochure or website — to determine classification.
Vague intended use statements create ambiguity that often results in queries from the MDA. Overly broad statements that cover more uses than the device actually supports can trigger a higher classification than necessary. Statements that are too narrow may not accurately reflect the device's true function — which becomes a problem if the device is later found to be used differently in the field.
The intended use statement must be accurate, specific, and internally consistent with every other part of the technical file you submit.
Not Updating Classification When the Device Changes
A device that was correctly classified and registered at Class B may no longer be Class B if the manufacturer subsequently adds a new indication, modifies the device design in a way that changes the risk profile, or incorporates software that performs a clinical function.
Post-market changes that affect intended use, device function, or risk level may trigger the need for a reclassification assessment and potentially a new or amended registration. Manufacturers who assume their existing registration covers modified versions of their device often discover the problem during a post-market audit or when the MDA queries a renewal submission.
What Happens When a Device Is Misclassified
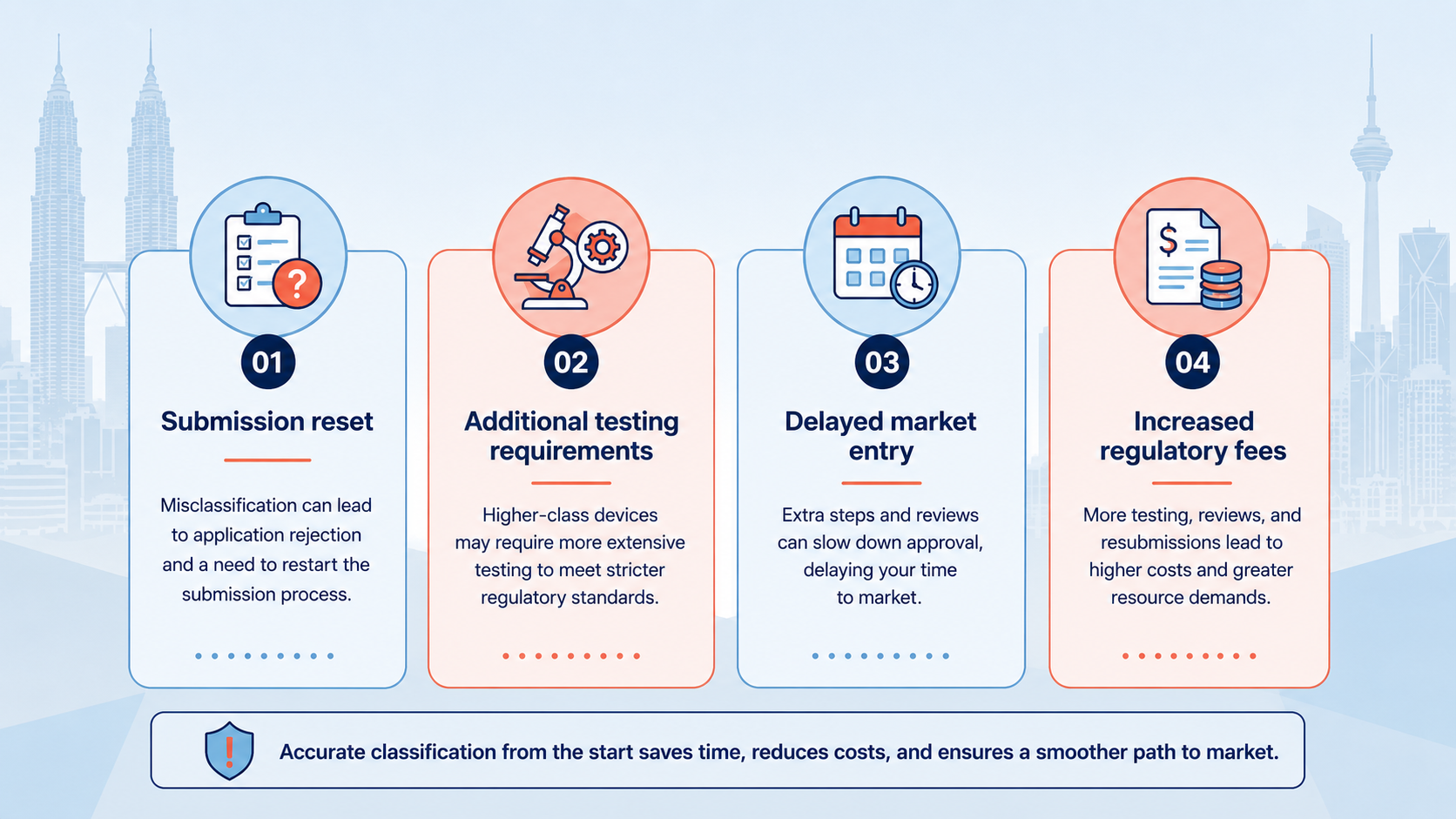
When the MDA determines during review that a device has been incorrectly classified, it issues a reclassification request. This is not a minor administrative correction — the consequences are significant.
In most cases, a reclassification request requires the submission to be withdrawn and restarted under the correct class. Documentation prepared for the wrong class is typically not reusable without significant revision.
If the correct class is higher than originally submitted, new testing evidence — such as conformity assessment, clinical data, or electrical safety testing — may be required before a new submission can be made.
A reclassification request typically adds a minimum of several months to the approval timeline. For products tied to distributor contracts or hospital procurement cycles, this delay can result in missed selling windows that cannot be recovered.
Re-submitting under a different class involves additional MDA submission fees, conformity assessment costs, and consultant time — all of which could have been avoided with correct classification from the start.
How to Confirm Classification Before You Submit
The most reliable way to avoid classification mistakes is to treat classification as a formal, documented exercise — not an assumption. Before any documentation is prepared, the following should be confirmed:
Classification Support from TT Medical
TT Medical works with manufacturers at the classification stage before any documentation is prepared. Getting this step right is how we help our clients avoid the delays, additional costs, and rework that come from classification errors discovered during MDA review.
Final Thoughts
Classification mistakes in medical device registration rarely look serious at the outset. They tend to surface during MDA review — at which point the cost of correcting them is far higher than the cost of getting them right from the start.
The common thread across all of the mistakes covered in this guide is the same: classification based on assumptions rather than a deliberate, documented assessment of intended use and the applicable rules. Fixing that one habit — before documentation begins — is the most effective thing any manufacturer can do to protect their registration timeline.
If you're unsure how your device should be classified, or if it falls into any of the grey areas covered here, speak to our consultancy team before you begin. A classification review takes a fraction of the time a reclassification request will cost you.
FAQ