Technical documentation for MDA registration in Malaysia is prepared in the Common Submission Dossier Template (CSDT) format, governed by MDA guidance document MDA/GD/0008. It must cover seven core elements: an executive summary, the Essential Principles conformity checklist, device description, design verification and validation summary, clinical evidence summary, labelling, and manufacturer information. Every section must be addressed, every referenced document submitted in full, and every certificate signed, dated, and within its validity period.
If you've confirmed your device classification and you're ready to start preparing your submission, the next — and often most time-consuming — step is the technical documentation itself. For Class B, C, and D devices, this means building a complete CSDT (Common Submission Dossier Template).
The CSDT isn't a form you fill in once and forget about. It's a structured technical file that has to demonstrate, with evidence, that your device meets every applicable Essential Principle of Safety and Performance (EPSP) under the Medical Device Act 2012 (Act 737). Get the structure right and the rest of your submission moves smoothly. Get it wrong, and you'll spend months responding to MDA queries.
This guide walks through exactly how to prepare your technical documentation — section by section — based directly on MDA's own guidance document MDA/GD/0008. For the full list of supporting documents required alongside the CSDT, see our complete MDA document checklist.
Key Takeaways
- The CSDT is the standard technical documentation format for Class B, C, and D devices, governed by MDA/GD/0008.
- It must cover seven core elements — executive summary, EPSP checklist, device description, design verification and validation, clinical evidence, labelling, and manufacturer information.
- Every referenced supporting document must be submitted in full, legible, and within its validity period.
- All certificates and reports must be signed off and dated by the person issuing the document.
- Where a CSDT element does not apply to your device, a written justification for non-applicability is required.
- The depth and detail required scales with device classification — higher risk classes need more comprehensive evidence.
- For re-registration, no changes to existing CSDT information are permitted unless approved by the MDA via a change notification.
What Is the CSDT and Why Does It Exist?
The CSDT format is based on the GHTF guidance document "Summary Technical Documentation for Demonstrating Conformity to the Essential Principles of Safety and Performance of Medical Devices" (STED). Malaysia adopted this internationally aligned structure to give manufacturers a single, consistent way to demonstrate conformity — rather than reinventing documentation requirements market by market.
The underlying goal, as stated in MDA's own guidance, is to strive for the least burdensome means of demonstrating conformity to the Essential Principles across all device classes. That doesn't mean the CSDT is simple — it means the format itself is designed to avoid unnecessary duplication once you understand how to structure it correctly.
The Seven Core Elements of the CSDT
Every CSDT must address these seven elements in sequence. This is the backbone structure prescribed by MDA/GD/0008 — understanding what goes into each one is the foundation for preparing a submission-ready dossier.
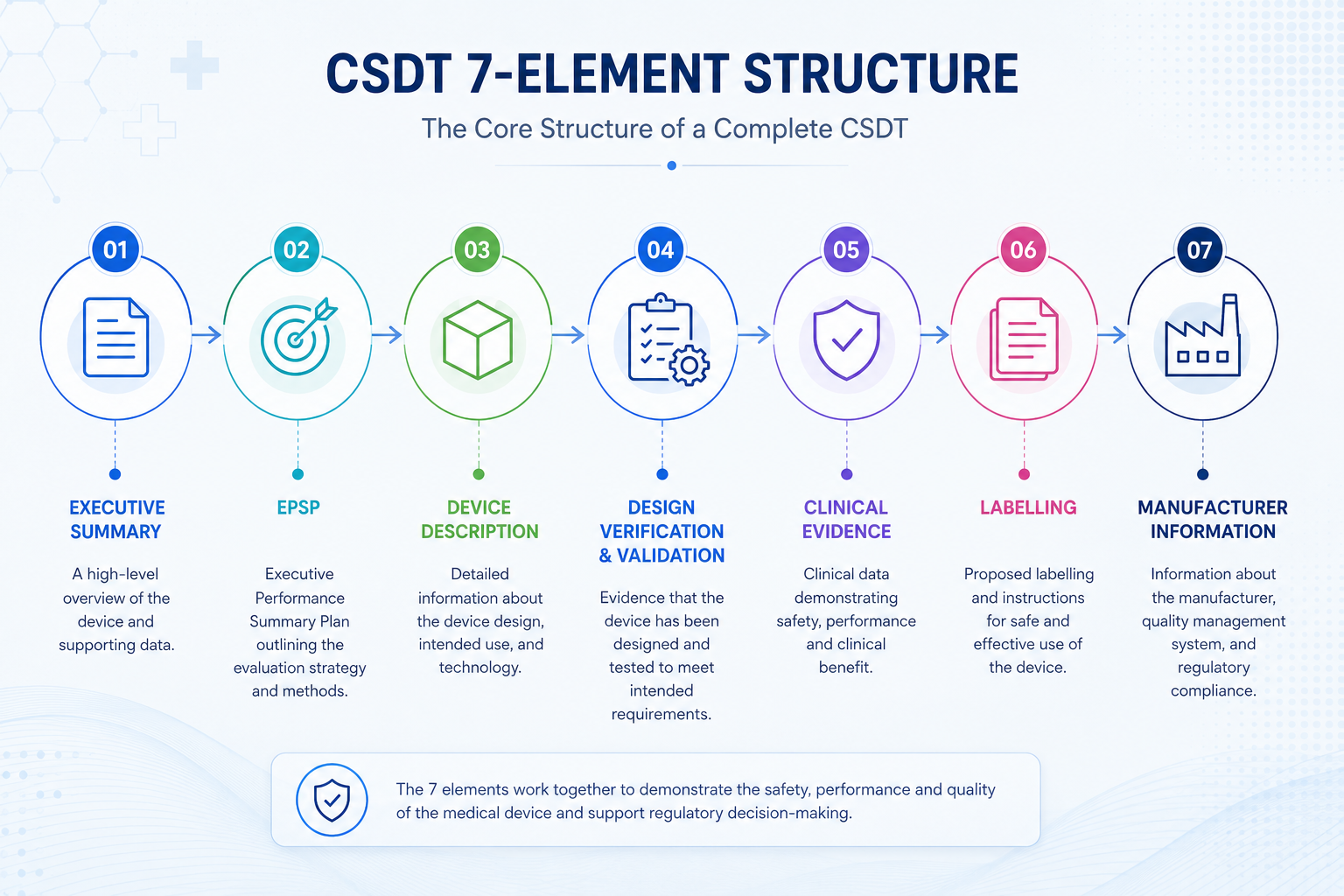
An overview of the device, its intended use and indications, any novel features (e.g. nanotechnology), and a synopsis of the CSDT content. Also includes commercial marketing history, prior regulatory approvals across reference agencies, and copies of certificates or approval letters from each reference agency.
The Essential Principles Conformity Checklist — provided alongside supporting documentation. Each applicable principle must identify the standard used (full title, identifying numbers, date, and issuing organisation) or, where internal standards are used, equivalent detail.
A complete description of the device's key functional elements — parts, components, software if applicable, formulation if combined with a drug component, and labelled pictorial representations where appropriate. Includes a list and full characterisation of materials making direct or indirect contact with the body.
Pre-clinical physical testing (mechanical, electrical safety, accelerated aging), software validation studies where applicable, and specific considerations for devices containing biological material. This is the evidence backbone supporting your safety and performance claims.
Clinical experience with the same or similar devices, comparing clinical, technical, and biological characteristics — including justification of clinical impact for any differences. Real-world data, device registries, and post-market clinical follow-up (PMCF) data should be included where applicable.
All labels, packaging, and Instructions for Use (IFU) — covering procedures, methods, frequency, duration, quantity, and preparation needed for safe use. Where possible, safe-use instructions should appear on the device itself and/or its packaging, not only in a separate manual.
Manufacturing site details, quality management system certification (ISO 13485 or recognised equivalent), and authorised contact person details. This section anchors the CSDT to a verifiable, accountable manufacturing organisation.
A risk analysis aligned with ISO 14971 runs throughout the CSDT — not as a standalone section but as supporting evidence woven through device description, design verification, and clinical evidence. See our active vs non-active devices guide for how device type affects which risk evidence applies.
How Much Detail Does Each Section Need?
MDA's own guidance is explicit on this point: the depth and detail of information in the CSDT depends on factors including device classification, the complexity and novelty of the technology, and the level of risk associated with the device. A higher-risk device needs more comprehensive evidence in every section — not just the clinical evidence section.
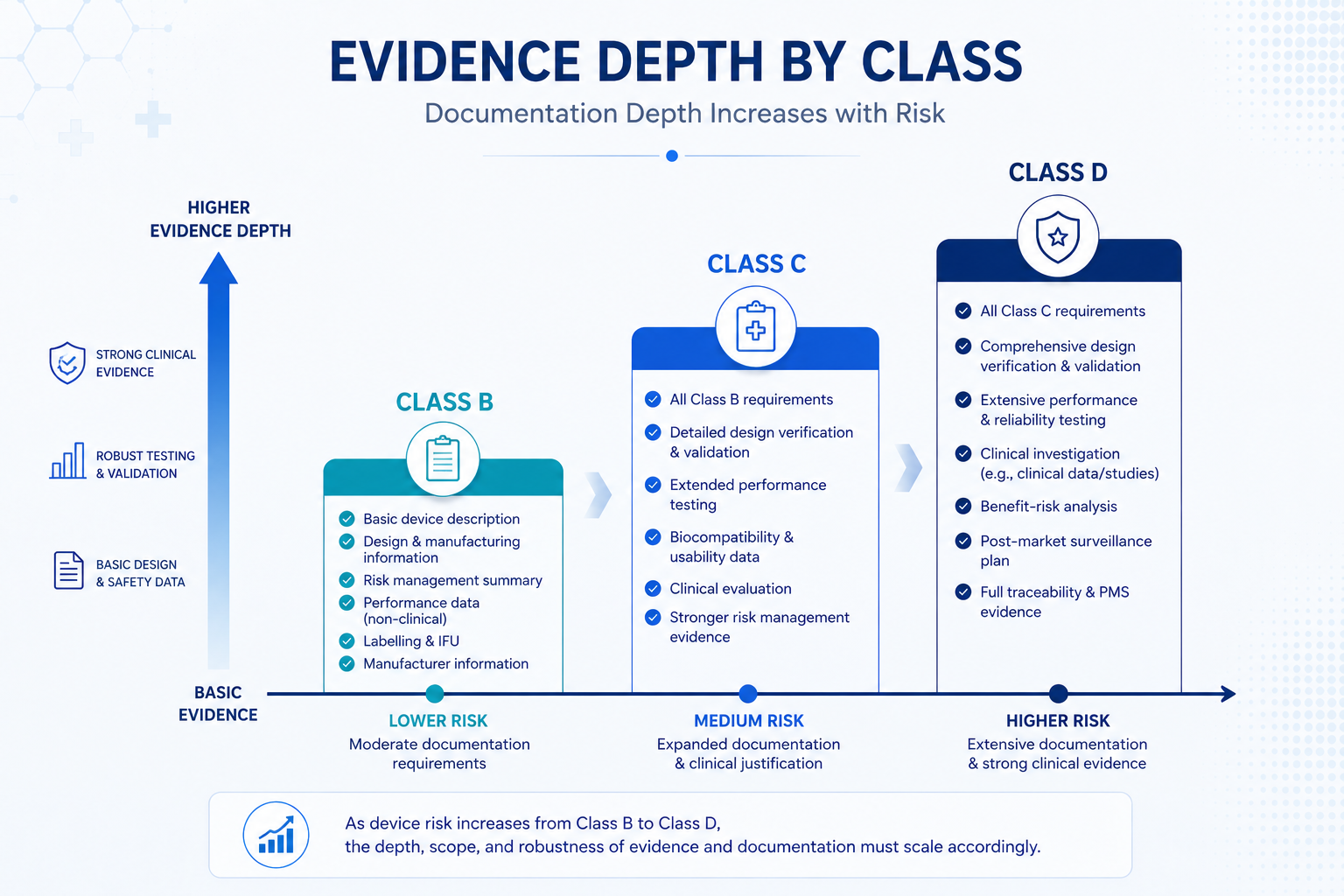
A clinical evidence summary referencing similar marketed devices is typically sufficient, alongside standard design verification testing. The EPSP checklist still needs full cross-referencing, but the supporting evidence package is lighter than higher classes.
A full clinical evaluation report is expected rather than a summary, supported by systematic literature review or clinical investigation data. Biocompatibility, software, and electrical safety evidence become mandatory where applicable to the device type.
Full clinical investigation data or comprehensive systematic literature review with extended long-term follow-up is required. Every section of the CSDT — risk analysis, design verification, manufacturer QMS evidence — must reflect the highest level of regulatory scrutiny given to life-sustaining or life-supporting devices.
Three Rules That Apply to Every CSDT Submission
Regardless of device class, MDA's guidance documents repeat three requirements consistently across every section of the CSDT. These are not suggestions — they are conditions for an acceptable submission.
Where a supporting document is referenced within the CSDT, all pages of that document must be submitted — not excerpts or summaries. Partial submissions are treated as incomplete and will trigger a query.
Every certificate or report submitted must be clearly legible and within its stated validity period at the time of submission. Expired certificates — even if valid when the testing was performed — are not accepted.
Every certificate or report submitted must be signed off and dated by the person issuing the document. An unsigned or undated document — regardless of how complete its content is — will be rejected at first review.
One additional structural requirement applies throughout: where a CSDT element does not apply to your specific device, you must provide a written justification for the non-applicability. Leaving a section blank without explanation is treated the same as an incomplete submission.
How to PrepareHow to Prepare Your CSDT: A Practical Workflow
Everything else in the CSDT — classification, EPSP checklist, clinical evidence scope, labelling — flows from how you describe the device's intended use. Get this consistent and accurate before drafting any other section.
Build the EPSP checklist early. For each applicable principle, identify the standard you'll reference (full title, number, date, issuing body) before drafting the supporting evidence — this avoids rework later.
Gather existing test reports, design verification data, clinical evidence, and QMS certificates. If you already hold CE marking or US FDA clearance, much of this evidence can be reused — but verify it's current and complete.
- Pre-clinical and bench testing reports
- Software validation documentation (if applicable)
- Clinical evaluation data or literature review
- ISO 13485 certificate and manufacturing site details
Work through the executive summary, EPSP checklist, device description, design verification summary, clinical evidence summary, labelling, and manufacturer information — in that order. Cross-check each section against the intended use statement for consistency.
Check every reference in the CSDT points to a document that is included in full, legible, signed, dated, and within validity. This is the single highest-impact review step before submission to your CAB.
The CAB reviews the CSDT against the EPSP checklist and verifies compliance with essential safety and performance principles before issuing its certificate and assessment report — required before MDA submission.
Common CSDT Preparation Mistakes
The intended use described in the executive summary must match the device description, labelling, IFU, and clinical evidence sections exactly. Drift between sections is one of the most common triggers for MDA queries.
Referencing a test report but only submitting a summary page, rather than all pages of the document, is treated as an incomplete submission regardless of how relevant the excerpt is.
If an element genuinely doesn't apply to your device, a written justification is required — not silence. Blank sections without explanation are treated as missing information.
Every certificate and report must be signed, dated, and within its validity period at submission. This applies to ISO 13485 certificates, test reports, and prior regulatory approvals alike.
A clinical evidence summary that would satisfy a Class B submission is insufficient for Class C or D. Matching evidence depth to classification is essential — not optional.
For re-registration, no changes to existing CSDT information are permitted unless approved by the MDA through a formal change notification. Updating content informally can invalidate the re-registration submission.
CSDT Preparation Support from TT Medical
Preparing a CSDT that satisfies both the CAB and the MDA on the first attempt requires close attention to structure, consistency, and evidence completeness. TT Medical works with manufacturers to build CSDTs from the ground up — or to review and tighten an existing draft before it goes to the CAB.
Final Thoughts
The CSDT is the most demanding document in your registration package — but it's also the most predictable, because the MDA has published exactly what it expects. Following the seven-element structure, matching evidence depth to your device class, and respecting the three non-negotiable rules around completeness, validity, and signatures will take you most of the way to a clean submission.
Where manufacturers most often run into trouble is not the content itself, but consistency — between the intended use statement, the labelling, the clinical evidence, and every cross-reference in between. If you'd like a second set of eyes on your CSDT before it goes to the CAB, speak to our consultancy team.
FAQ